By Carol Beuchat PhD
This week I had the pleasure of speaking at the Bernese Mountain Dog national specialty being held in Portland, Oregon. They asked me to talk about genetics, and my original plan was to discuss some of the new tools available to breeders that can help them make the best possible breeding decisions.
I thought I would start with a brief summary of the genetic information available about Berners, but I found there was much more published data than I anticipated. In fact, I had so much information to share about the breed, and there were so many questions as we went along, that I ran over my 2 hour time slot by a half hour and we continued the discussion for another 3 hours over lunch!
For the benefit of those that missed the seminar, and also for those that attended that asked for a summary, I've copied most of the figures I used and added brief explanations.
I thought I would start with a brief summary of the genetic information available about Berners, but I found there was much more published data than I anticipated. In fact, I had so much information to share about the breed, and there were so many questions as we went along, that I ran over my 2 hour time slot by a half hour and we continued the discussion for another 3 hours over lunch!
For the benefit of those that missed the seminar, and also for those that attended that asked for a summary, I've copied most of the figures I used and added brief explanations.
Genetic Relationship to Other Breeds
In this figure published in 2015, the Bernese Mountain Dog clusters with the Greater Swiss Mountain dog (highlighted).
In this figure published in 2015, the Bernese Mountain Dog clusters with the Greater Swiss Mountain dog (highlighted).
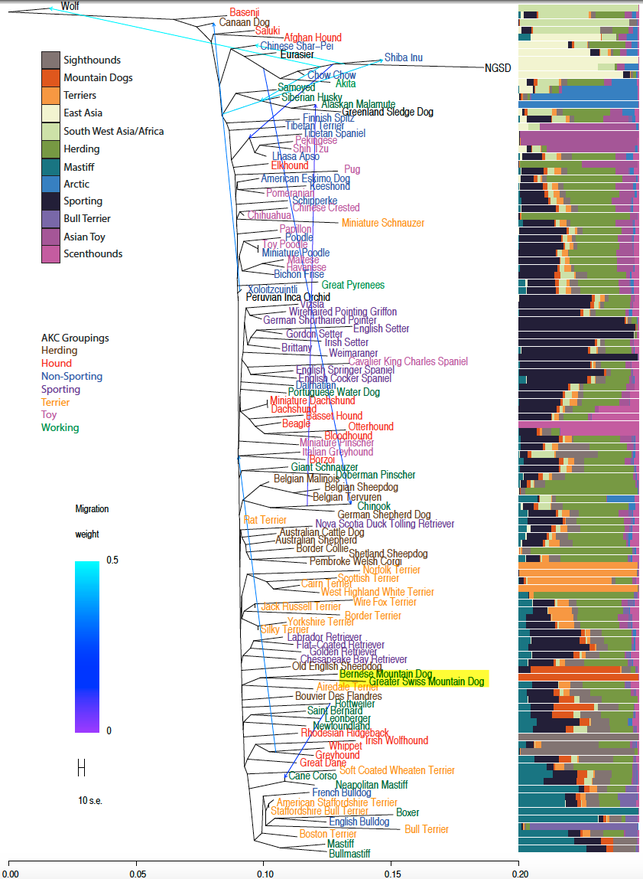
Shannon et al 2015.
A new study of the genetic relationships among dog breeds was just published (yesterday!), so I didn't have this figure in my talk but it's worth including here as the most up-to-date information (Parker et al 2017). Bernese Mountain Dog is at the red arrow.
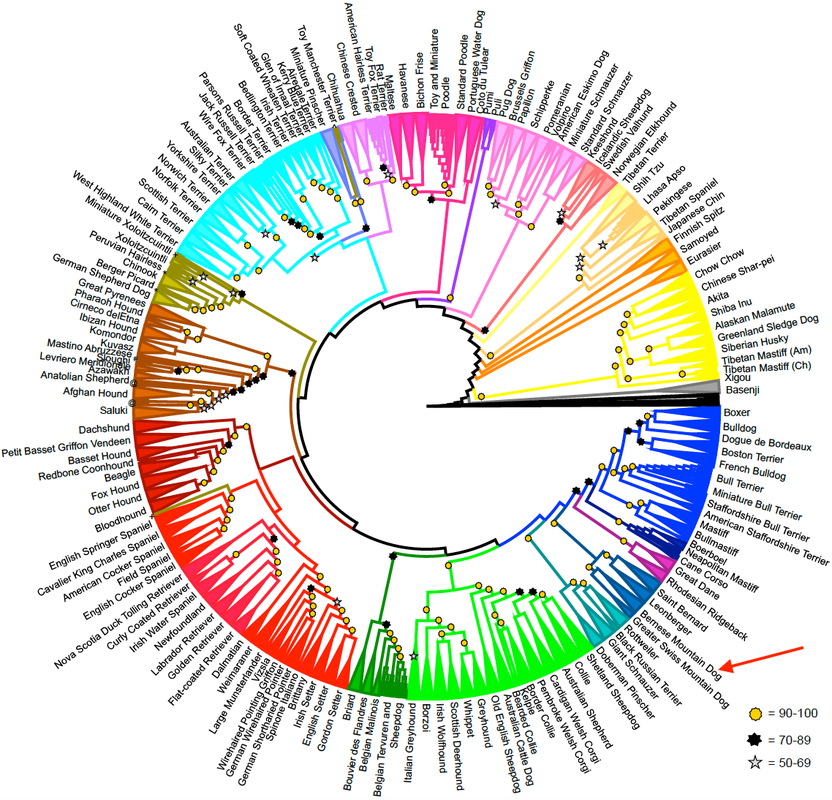
Breed History
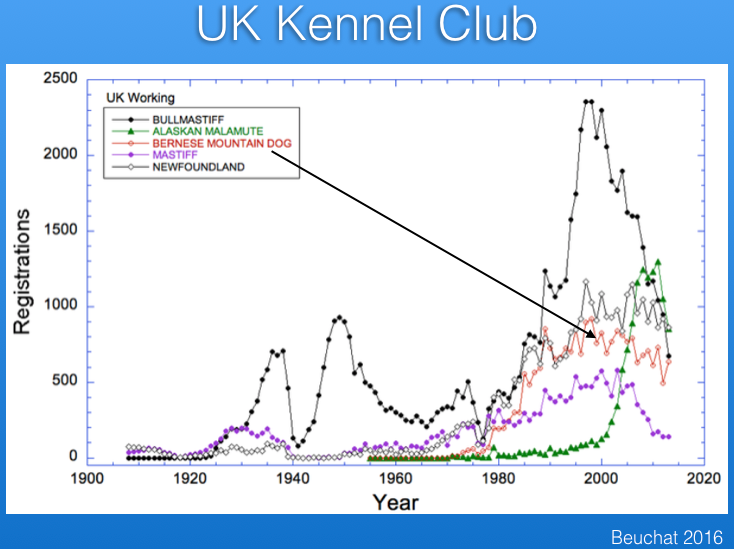
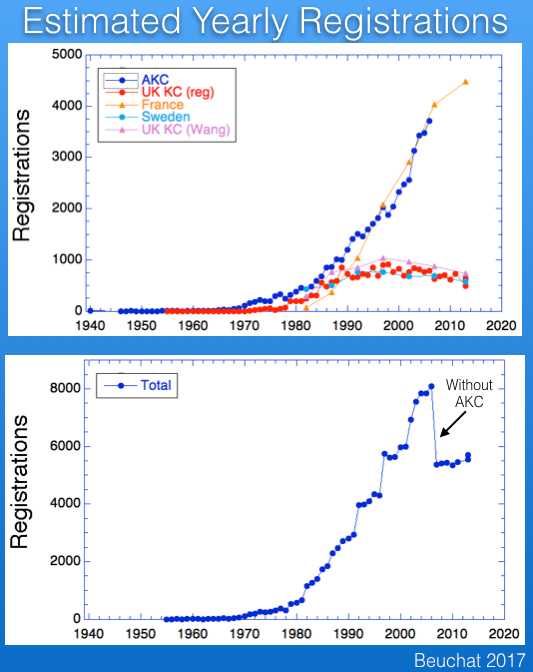
There are registration records available for several countries. Below are the data from the UK Kennel Club for number of Berner registrations per year, together with those for several other breeds for comparison. The breed became more popular after about 1980, and although registrations have declined a bit in the last 10-15 years, the breed is faring much better than some that have experienced very significant drops in registrations (e.g., Bullmastiff).
A recent study (Wang et al 2016) has published data for registrations from France and Sweden (and the paper included the UK as well), which I have graphed below. Note that these data are reported over 5 year blocks, so the numbers are not comparable to the yearly registration figures for the UK data above.
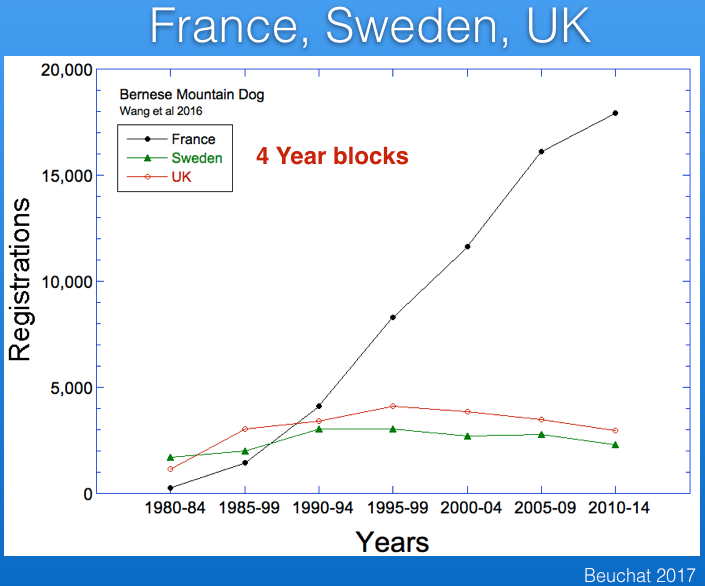
I wanted to compare the data from these countries, so I divided the numbers from the Wang et al study by five and combined with the UK data and also data for the US from AKC. These data are graphed below.
The striking thing about these data is the popularity of the breed in France, which has registration figures comparable to those in the US and about five times greater than the UK and Sweden.
I also added the registrations for a total from these four countries, which is plotted below. The AKC data run only through 2006, which is why the total drops suddenly ("without AKC").
Inbreeding
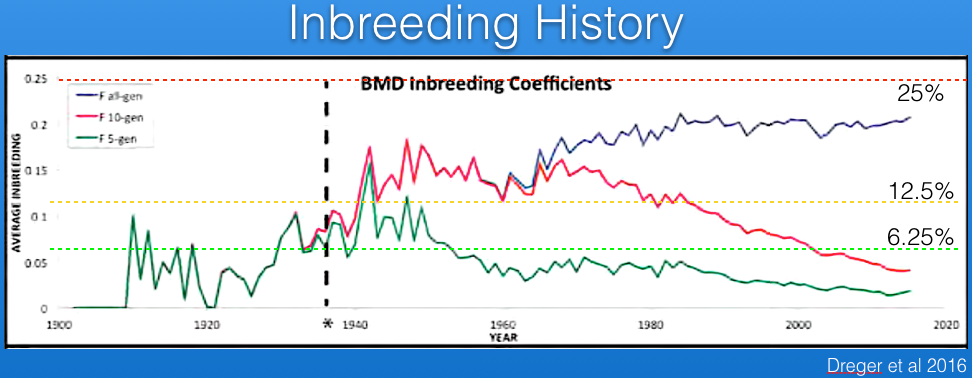
Parker et al (2017) have analyzed the Berner-Garde pedigree database and produced the graph below of coefficient of inbreeding. The blue line uses all of the data in the database back to founders for the calculations, while the red and green lines use only the previous 10 and 5 generations, respectively. The current average inbreeding estimated from the pedigree data back to founders is about 20%.
Using only 10 or 5 generations of data, the calculated inbreeding coefficient is less than using the full pedigree because it reflects only the inbreeding that occurred over those generations. This is because there is no information about the earlier history that goes into the calculations. Notice also that missing pedigree information or errors in the database will result in an underestimate of inbreeding coefficient because earlier inbreeding is not taken into account.
The inbreeding coefficient is the probability of inheriting two copies of the same allele from a parent. In the case of a recessive mutation, the inbreeding coefficient is therefore also an estimate of the risk of producing a genetic disorder. For example, a full-sib cross produces offspring with an average inbreeding coefficient of 25%, and the risk of inheriting two copies of the same mutation is also 25%. Of course, if you only use a few generations of pedigree information behind your dog of interest, you will calculate a much lower inbreeding coefficient but the actual probability of inheriting two copies of the same allele is still the same. It's important to use the inbreeding coefficient properly (i.e., full pedigree data) and understand how the statistic should be interpreted.
(Breeders should take advantage of the free online course "COI Bootcamp" available through ICB.)
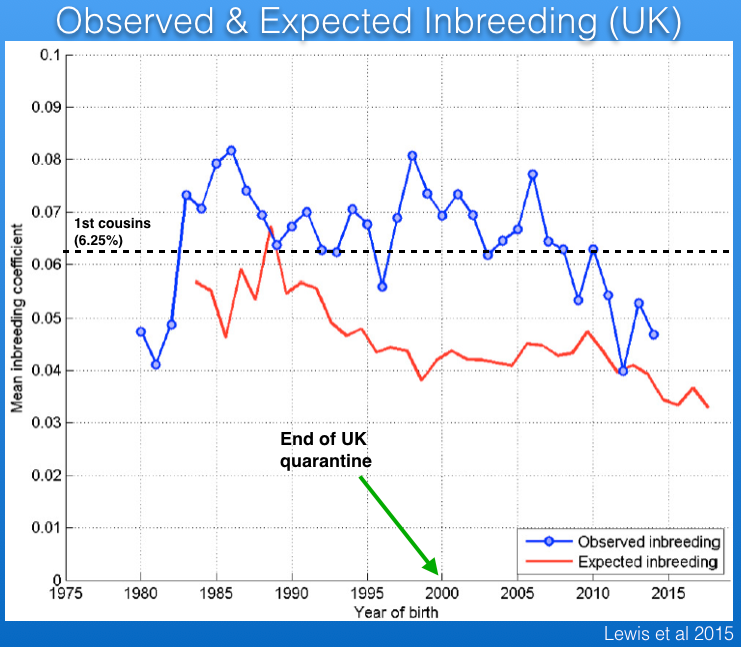
There are data for the history of inbreeding in the Bernese Mountain Dog in the UK from 1980 to the present that were published by Lewis et al (2015). Note that these data are not comparable to those just described because the UK pedigree analysis does not take into account inbreeding that occurred prior to 1980. (Their digital records only go back that far.) Consequently, these data underestimate the actual inbreeding by an unknown magnitude. Furthermore, the restrictions for importation of dogs into the UK were lifted in 2000. Because the pedigree records for imported dogs go back only three generations, this graph gives the false impression that inbreeding is decreasing over the last 15 years or so.
One more point about this graph is the difference between observed (blue) and expected (red) inbreeding. The latter is what you would expect to see in an "ideal" population - i.e., randomly breeding animals with no selection and no migration in or out. When observed inbreeding is higher than expected inbreeding, that reflects breeding strategies that are pairing animals that are more closely related than the average in the population; i.e., breeders are preferentially inbreeding.
Inbreeding can be estimated from pedigree analysis, but this is a statistical prediction and can be wildly incorrect if the quality of the database is poor because of missing information and errors.
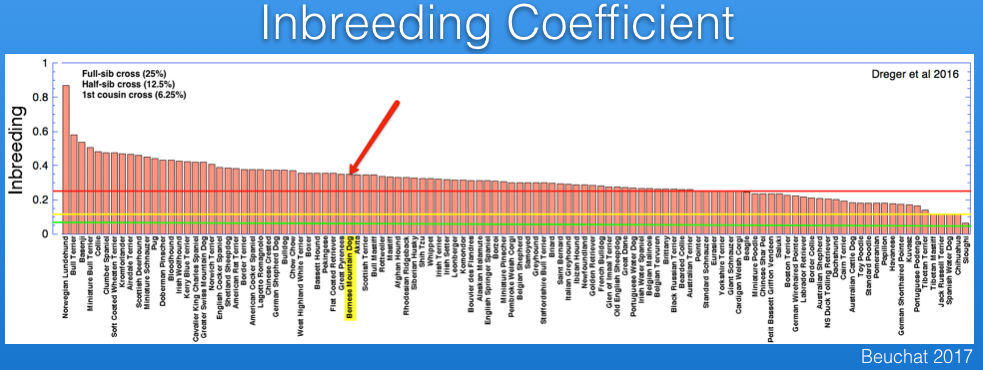
Inbreeding can also be estimated from DNA analysis that uses information about thousands of markers. The study by Dreger et al. provides information for inbreeding from DNA data, and I have graphed below. (You can also see these data and additional discussion in my blog post.)
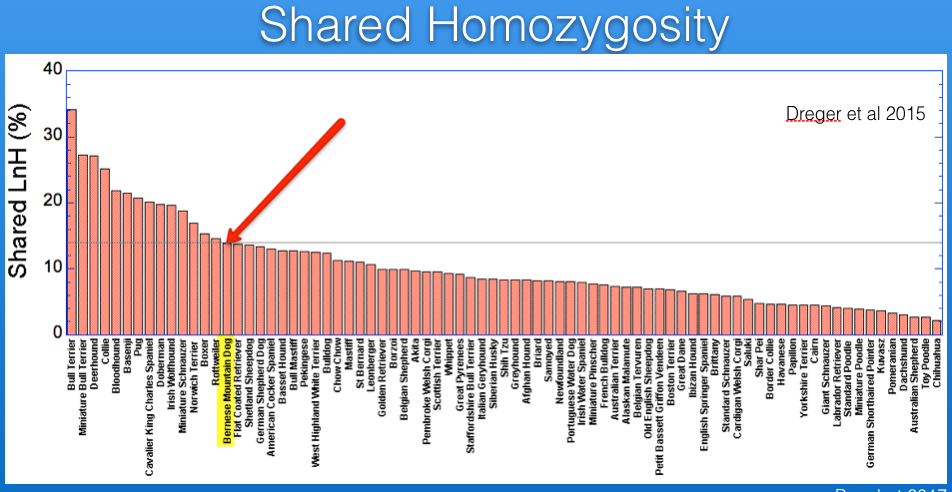
I have superimposed on this graph lines for the expected inbreeding levels from full-sib (25%, red), half-sib (12.5%, yellow), and first cousin (6.25%, green) crosses. The Bernese Mountain Dog is indicated by the arrow. From these data, it is evident that most of the breeds included in the study are more inbred than the offspring of a full-sib cross, and all but a handful are higher than you would expect from a half-sib cross.
The inbreeding coefficient of the Bernese Mountain Dogs in these data averages 35%.
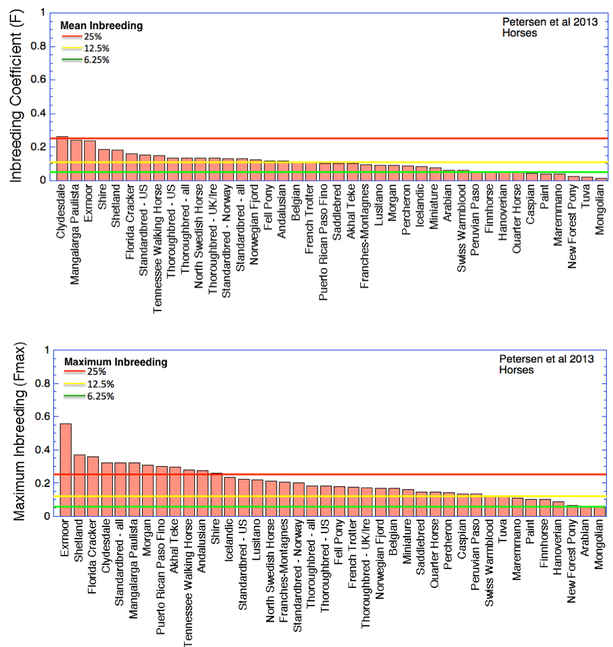
For comparison, I have also graphed comparable data for inbreeding in domestic horse breeds (the top graph is the mean, and the bottom is the maximum). You can see that horses are much less inbred than dogs, and most average less than what you would produce from a half-sib cross. The highest average inbreeding is just about 25% (for the Clydesdale).
These data show that extreme inbreeding is not necessary to produce domestic animal breeds that are recognizable in type and suited for their original function. No other domestic animal breeds approach the levels of inbreeding that are typical of purebred dogs.
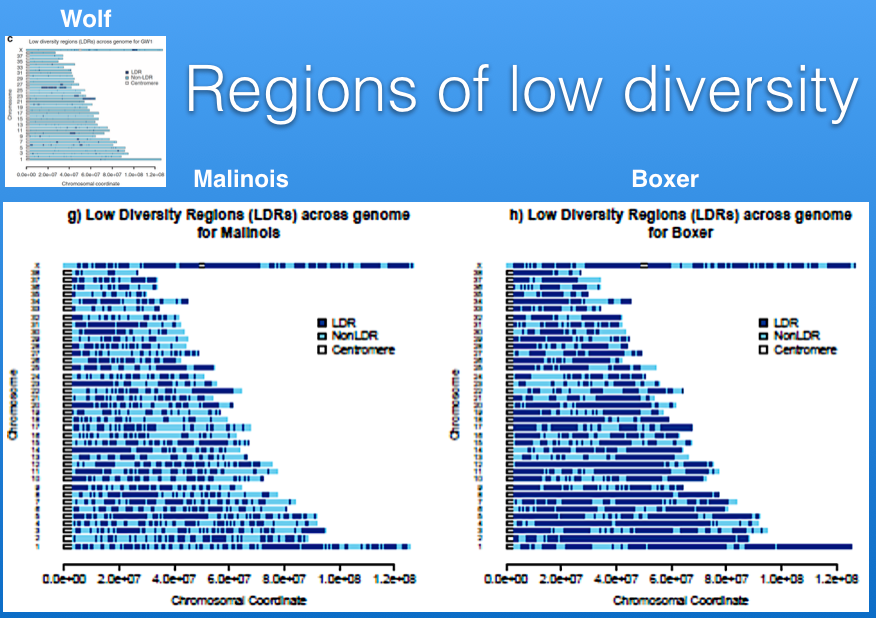
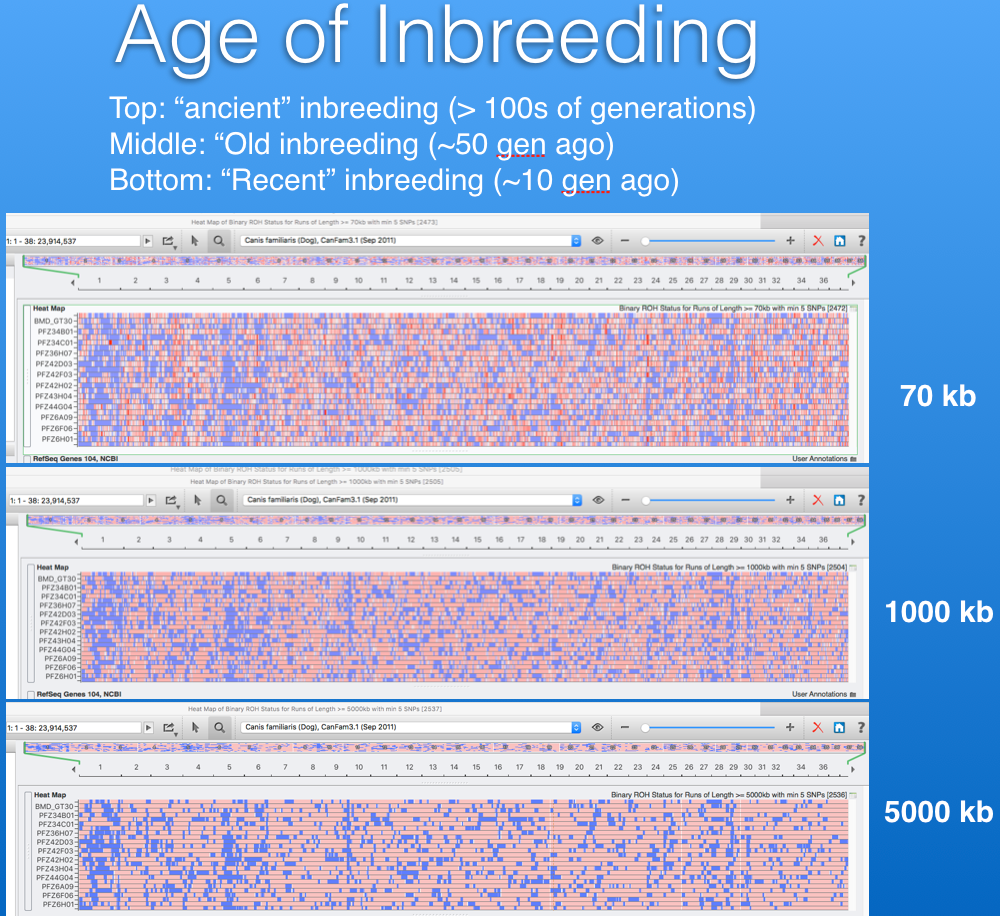
With the amazing DNA technology now, we can map regions of low diversity (high inbreeding or homozygosity) on the individual chromosomes. Below are some graphs depicting the 39 chromosomes in the dog (38 autosomal chromosomes plus the sex chromosomes. The regions of low diversity are in dark blue, and areas that are heterozygous are in light blue. The small graph in the header is of a wolf; you can see that most of the chromosomes are light blue. Below are two purebred dog breeds, the Belgian Malinois (left), and the Boxer (right). It's easy to see that both of the dog breeds have much more homozyosity than the wolf, and the Boxer has more than the Malinois.
We can also use the DNA data to determine areas of the chromosome that are homozygous in all individuals. You might expect this for areas of the chromosome that contain genes important to breed type. There could also be other regions that are homozygous in all dogs because of historical inbreeding. These regions of "shared" homozygosity are important because all future offspring will be genetically identical in those regions. Clearly, if genes of functional importance (e.g., for reproduction, kidney function, muscle contraction ) or for particular traits (color, behavior, size) are in these regions of shared homozygosity, selection will have no effect.
I have plotted below the data from Dreger et al. for shared homozygosity. The fraction of the genome with shared homozygosity in the Berner is about 14%. So, not only does the breed have a high average level of inbreeding, but about 14% of the homozygosity is common across individuals.
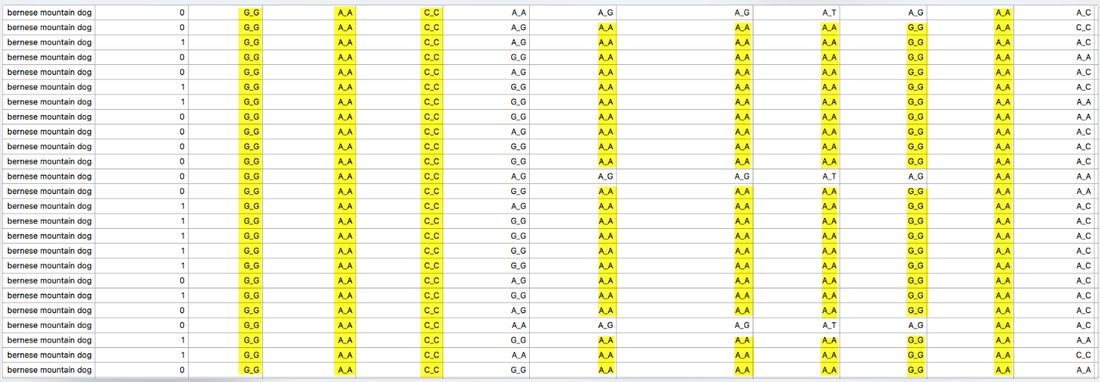
Below are two panels that display the regions of homozygosity in the genome of 12 Bernese Mountain Dogs (Beuchat, unpublished). Red regions are heterozygous and blue are homozygous. Each row is the depiction of all 38 chromosomes arranged end to end for a single dog (you can see the chromosome numbers in the strip just above the DNA scans).
It is easy to spot areas of shared homozygosity, which show as a vertical stripe of blue where the regions line up in most of the dogs. Elsewhere, you can see that blocks of inbreeding are scattered in different places for each dog.
In the lower panel, the image is zoomed in to a region on chromosome 5, and below the strip of DNA scans there is a panel that identifies the known genes in each region (too small to see, but indicated in blue and green). Each vertical line is a single SNP marker and you can see where that SNP is homozygous (red) or heterozygous (blue) for each dog and what gene it is associated with. This information could be used in making breeding decisions, e.g., to avoid matching up regions of homozygosity in the two parents, which would result in higher heterozygosity in the offspring.
This information can also be used to assess how old the inbreeding is. More recent inbreeding appears as longer blocks of homozygosity. Ancient inbreedingappears as shorter blocks, because with each generation there is a chance that a block will be broken up by the cross-over during meiosis. We can scan for short and long blocks of homozygosity and tell how much of the existing inbreeding is recent and how much is tens or even hundreds of generations ago (e.g., even before breed formation).
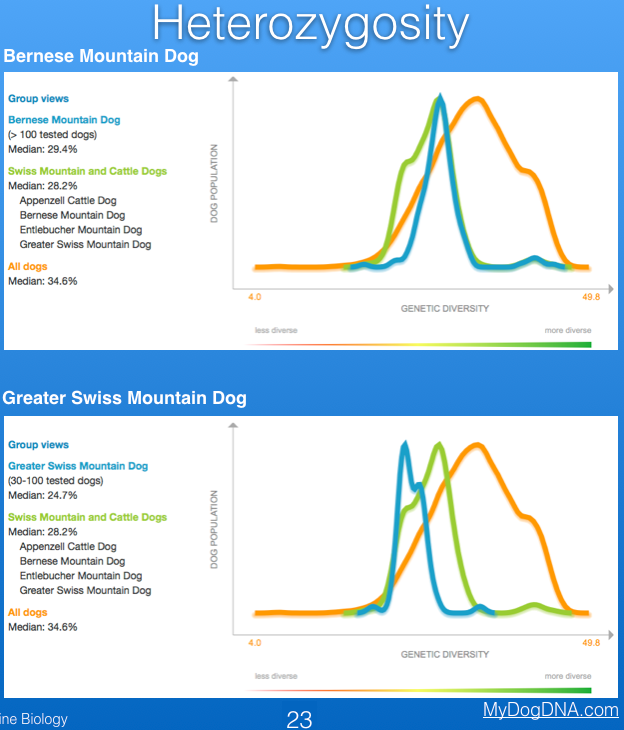
The Finnish company MyDogDNA reports information for breed-specific genetic diversity as heterozygosity, based on samples submitted to them for mutation and trait testing. The graph below shows that heterozygosity of the Bernese Mountain dog is very low (median = 29.4%), well below the "green zone" (see the strip below the graph indicating "more diverse" and "less diverse") and less than the median of all dogs (both purebred and mixed breed) in their database (orange line; 34.6%). Unfortunately, the closely related Greater Swiss Mountain Dog, which I have also included below for comparison, is even lower (median = 24.7%).
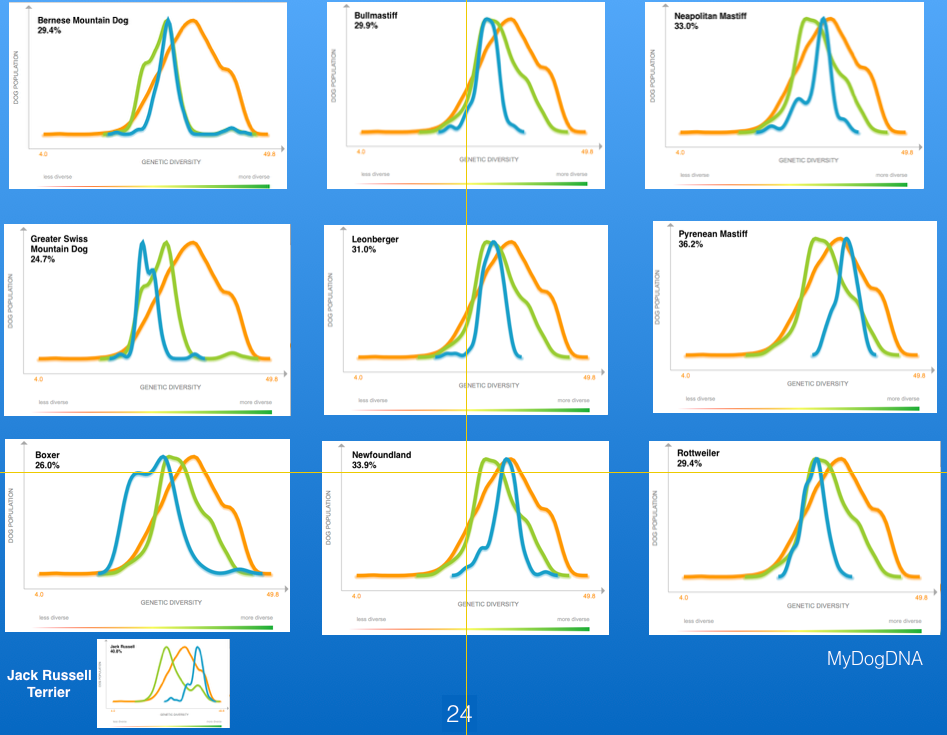
Below for comparison are the data for a number of working breeds, as well as the Jack Russell Terrier (lower left). Of these working breeds, the Pyrenean Mastiff has the highest heterozygosity, but it is still much lower than the Jack Russell Terrier. The lowest values for heterozygosity are in the Boxer, although the range in the breed is fairly wide.
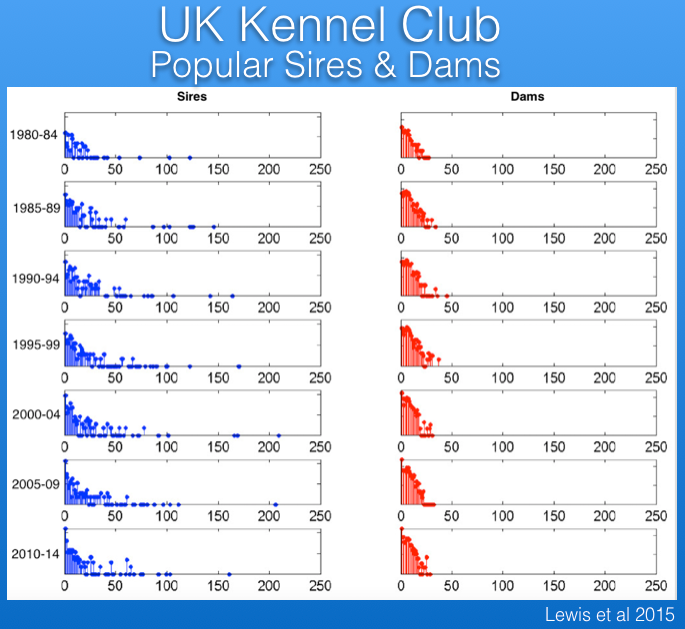
Popular Sires
Popular sires are responsible for loss of genetic diversity and also the wide dissemination of recessive mutations. The data from the UK Kennel Club reveal that there has been a serious problem with popular sires in Berners, with one or a few sires producing many times more offspring that most other males over the last 30 years.
For example, in the most recent period below (2010-2014), the average male produced < 25 offspring, while a single male produced > 150, and the data for previous time periods are similar.
In fact, only 5% of the males in the UK produce about 20% of all offspring, and about 90% of all offspring are produced by only 50% of the sires. (The data in red below should be labeled top 25%.)
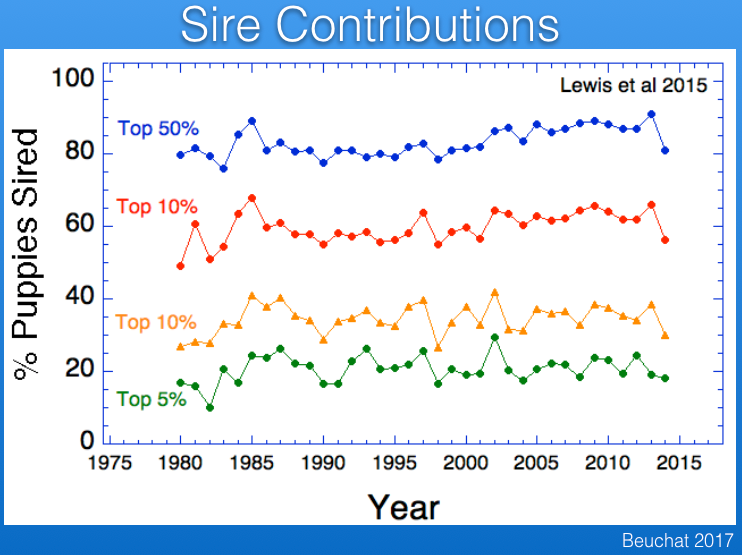
As is typical for most populations of purebred dogs, fewer males than females are used in breeding, and in Berners the difference is roughly a factor of two. Using fewer males than females means that there will be more half-siblings that share a sires. The result is that genetic disorders caused by recessive mutations are more likely to trace back to a sire than a dam.
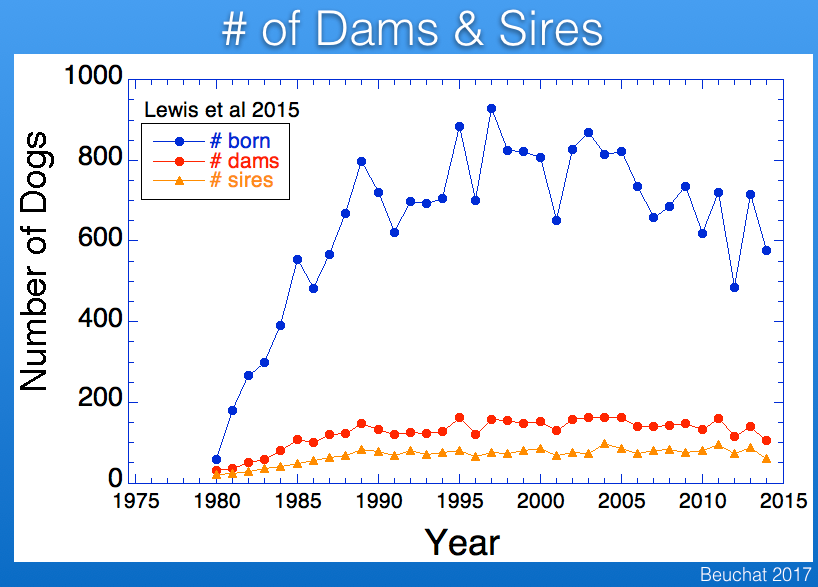
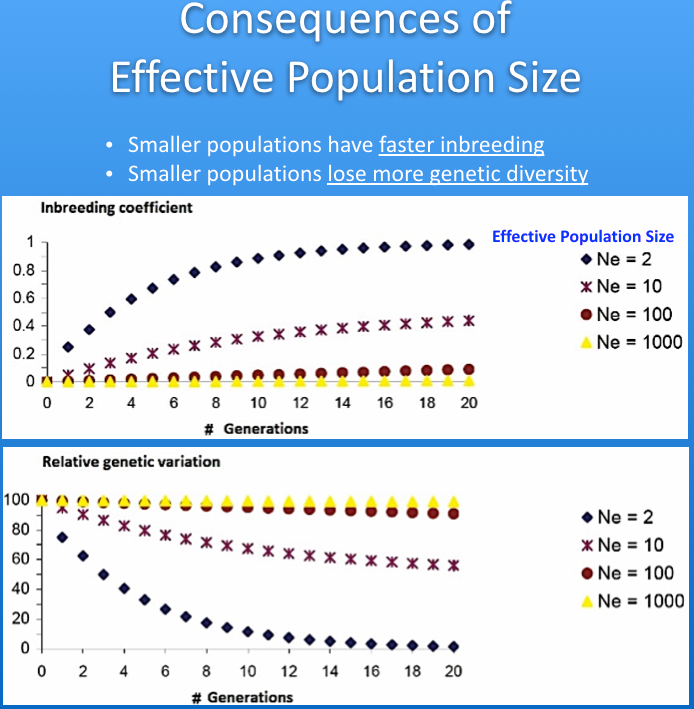
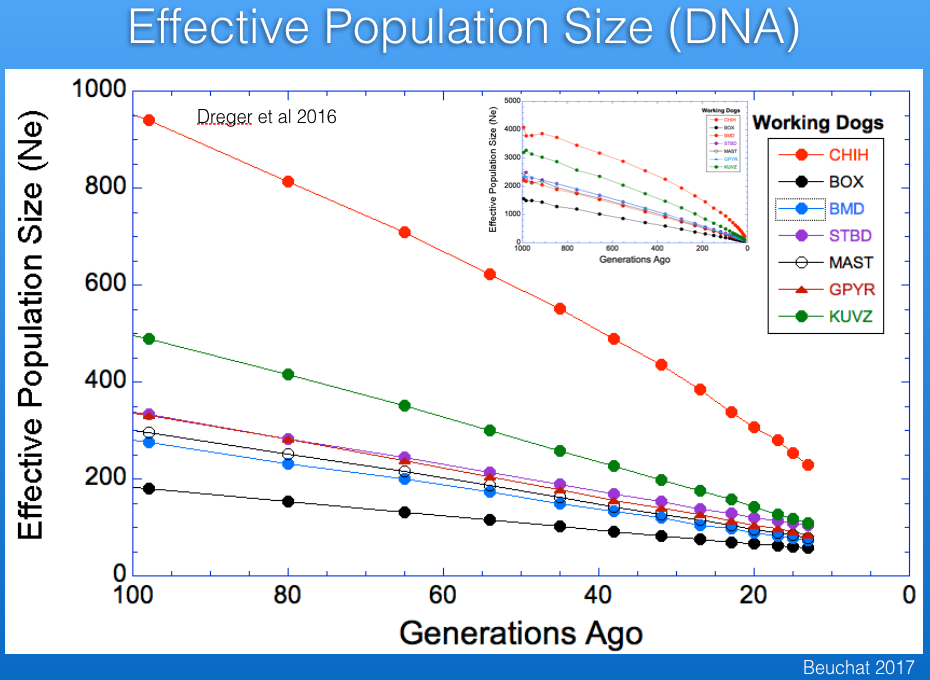
The imbalance in the ratio of males and females used for breeding causes inbreeding to increase faster than if the ratio of sires to dams was closer to one. This reduces the "effective population size" (Ne) of the breed. The graphs below show the influence of effective population size on the rates of increase in inbreeding and loss of genetic diversity. As a rule of thumb, when effective population size is 50, inbreeding will increase by 1% per generation (so, from 0% to 10% in 10 generations), and for Ne of 100 the rate of increase is 0.5% per generation. The higher the effective population size, the more genetically "stable" the population.
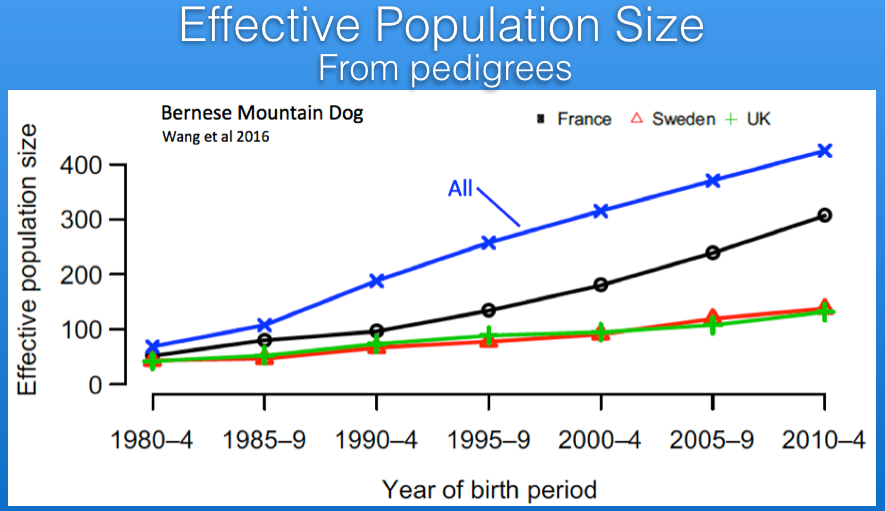
There are estimates of Ne based on pedigree databases for Berners in France, Sweden, and the UK. For Sweden and the UK, the current values of Ne are about 125, while that of France is roughly 300 (the latter in part because of the larger population size we saw earlier). If the populations in those three countries were combined, the estimate of Ne would be about 400. These estimates are from pedigree data so should be expected to overestimate the true Ne because of missing information or errors in the database.
Ne estimated from DNA data (probably mostly for the US population; Dreger et al 2016) is about 100 (blue in the graph below). You can see that it falls between the Boxer (black), the breed with the lowest Ne in this database, and the Chihuahua (red), the breed with the highest. As expected, Ne determined from DNA information is less than from pedigrees, but the populations are different, so it's hard to estimate the true effective population size for the breed.
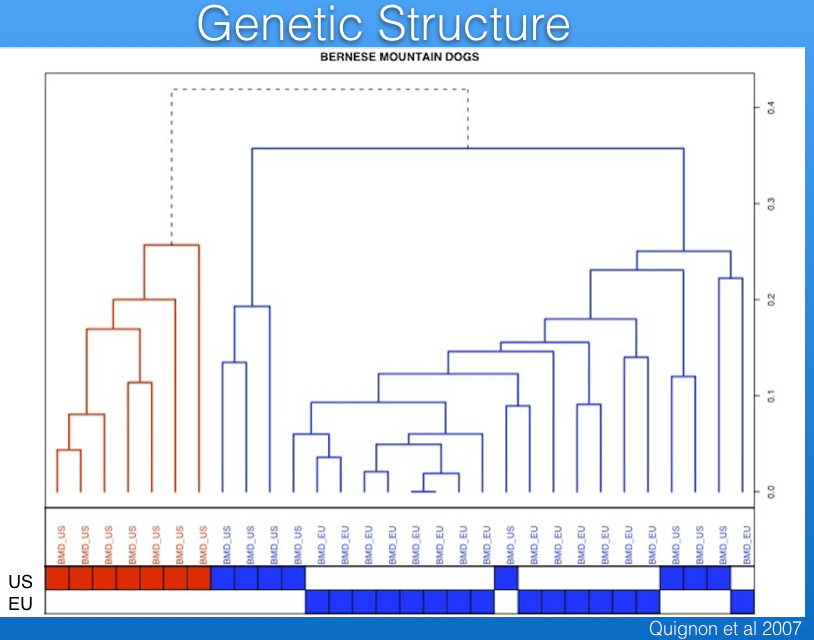
Genetic Structure
Populations that are separated geographically or that do not freely intermix for other reasons (e.g., selection protocols) will become less similar over time because of selection and genetic drift. These differences among subpopulations within a breed are referred to as "genetic structure".
For example, the data in the graph below show that there are multiple clusters of dogs that are genetically distinct; e.g., a cluster of related dogs in the US is separate from a larger cluster that is more diverse and includes dogs from both the US and European Union (EU).
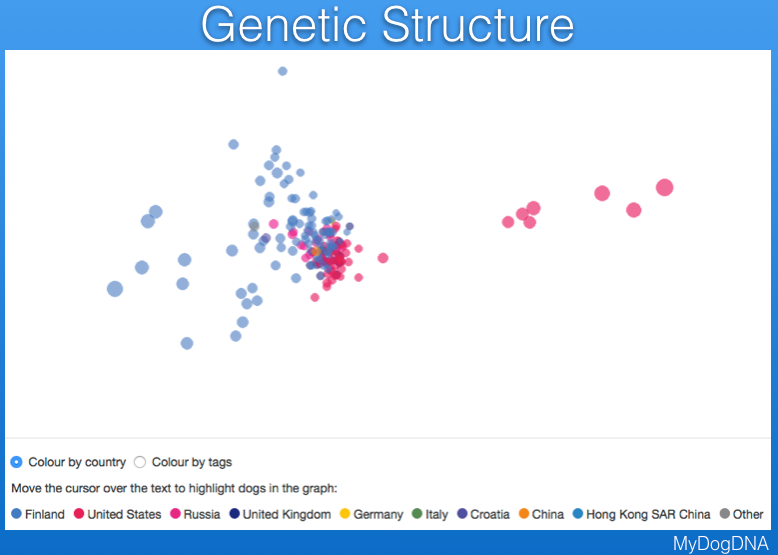
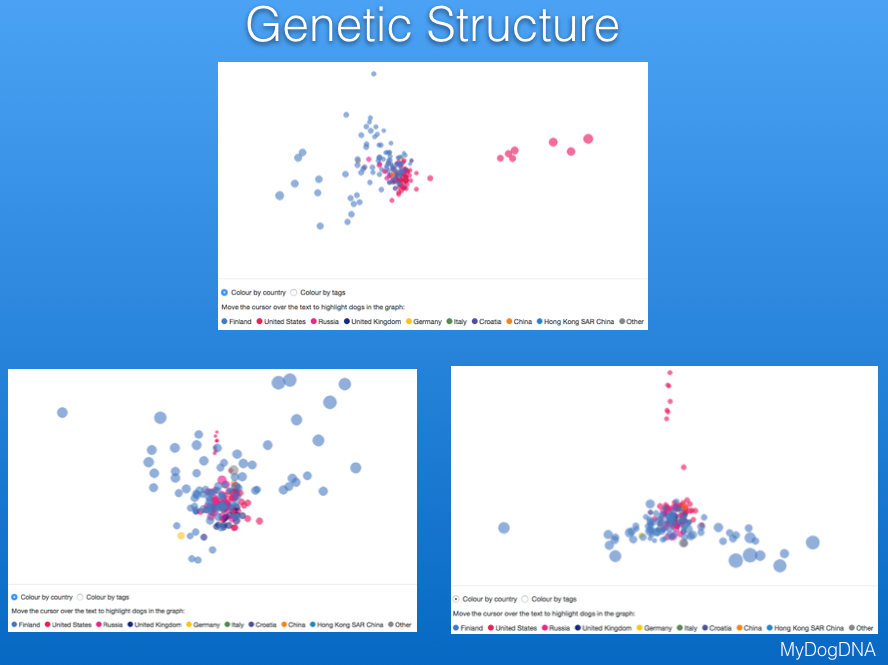
Another way to view the genetic structure in the breed uses "principal components analysis", which places each individual dog in a three-dimensional space, with distances between points reflecting genetic similarity. Below are data from MyDogDNA for Bernese Mountain Dog in which the data are color coded by country. They show that there is a main cluster of dogs from the US, but also some outliers in a string to the right, and a looser cluster of dogs from Finland (the light blue points to the left of the US cluster). The data are separated by country in the second plot below.
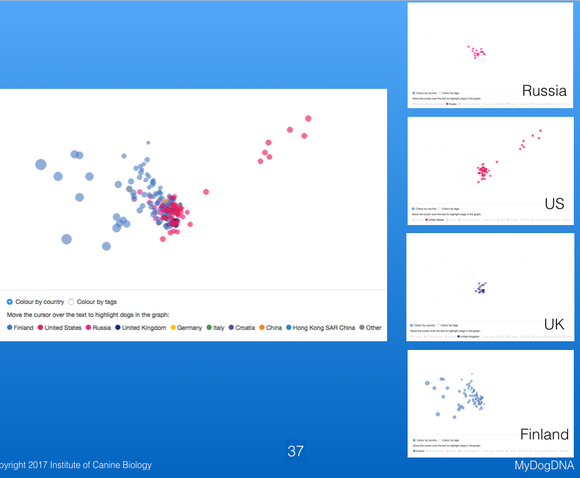
These plots are 3-D, and on the MDD website you can move the plot in space to view the relationships among points from different angles. These three plots are of the same data for Berners, with the difference being the viewing angle. These plots are much more informative for judging the genetic similarity or relatedness among animals than shuffling through piles of pedigrees looking for common ancestors. They are also really cool to play with; give it a try on the MyDogDNA website!
Size of the Gene Pool
The expression "size of the gene pool" refers to the breadth of genetic diversity in the population. The larger the gene pool, the higher the genetic diversity. We can standardize the estimate of genetic diversity by describing it as the number of unrelated animals that would produce the same genetic diversity as is seen in the population of interest. There are several versions of the statistics used by population geneticists for this.
Effective number of founders (fe) A breed might have 25 founders, but some of the original genetic diversity is invariably lost over time. The effective number of founders is an estimate of the number of founders that would produce the current genetic diversity of the population if all contributed equally to subsequent generations. This is a measure of the fraction of the genes contributed by the founders that still remain in the population.
Founder genome equivalent (fg) Founder genome equivalents of a population is that number of equally-contributing founders that would be expected to produce the same genetic diversity as observed in the current population if there is no random loss of founder alleles in descendants (e.g., through genetic drift).
Effective number of ancestors (fa) This is the minimum number of ancestors - which can be founders or not - needed to explain the genetic diversity of the current population. If there have been no population bottlenecks, fa will equal fe; the number and severity of bottlenecks will be reflected in the difference between fa and fe.
Founder genome equivalent (fg) Founder genome equivalents of a population is that number of equally-contributing founders that would be expected to produce the same genetic diversity as observed in the current population if there is no random loss of founder alleles in descendants (e.g., through genetic drift).
Effective number of ancestors (fa) This is the minimum number of ancestors - which can be founders or not - needed to explain the genetic diversity of the current population. If there have been no population bottlenecks, fa will equal fe; the number and severity of bottlenecks will be reflected in the difference between fa and fe.
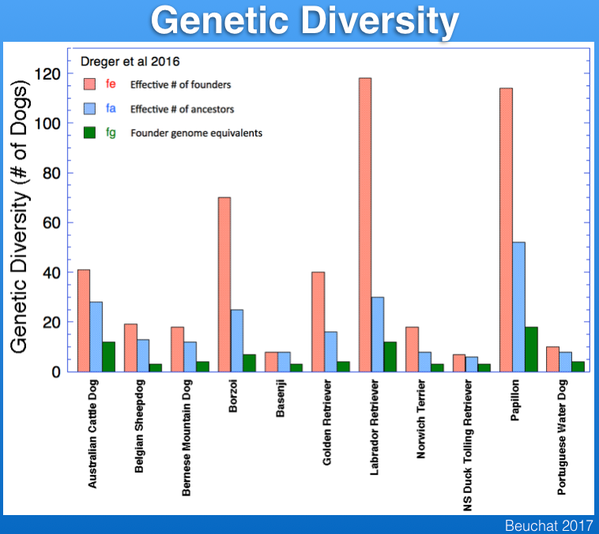
In the graph below are the diversity statistics for a few breeds. You can see that the estimates for genetic diversity of Berners are very low; fe is only about 20, and fg is only 4. This means that although there are thousands of dogs in the population, the breed has the genetic diversity of less than 20 dogs. These numbers were determined from analysis of the Berner-Garde pedigree database, and remember that if there are any missing data these estimates of diversity will be overestimates of the true value. For a genetically-stable, sustainably breeding population of animals, fe should be 50-100. To rescue a population at risk of extinction, the bare minimum for fe is about 20.
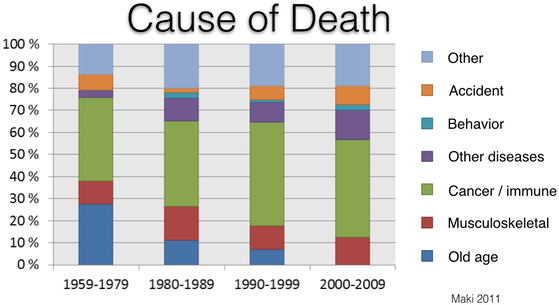
Cancer
About 60% of Bernese Mountain Dogs die of cancer (Shearin et al 2012). Histiocytosis is the most common cancer (17%) and is the focus of recent research initiatives, but it is only one of many. The incidence of cancer in Berners is among the highest of any breed and it kills them at a much younger age than most (typically 6-8 years old).
Efforts to identify the genetic cause(s) of cancer in Berners have come up empty, although histiocytosis is associated with a mutation in a tumor suppressor gene (CDKN2A; Shearin et al 2012).
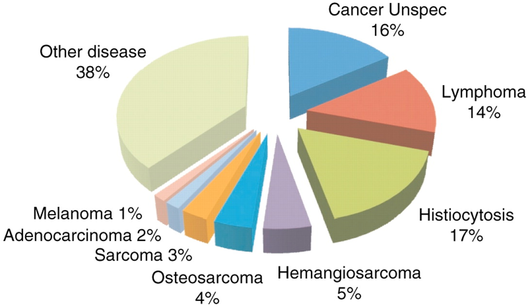
Shearin et al 2012
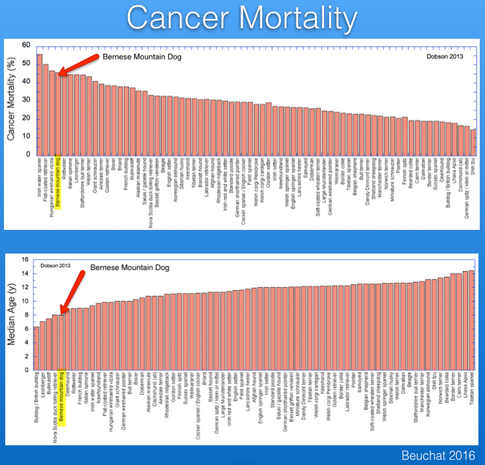
Over the last 50 years or so, the number of dogs dying of old age has dropped essentially to zero. Despite the efforts of breeders to reduce the incidence of cancer, however, it still remains the most significant cause of death.
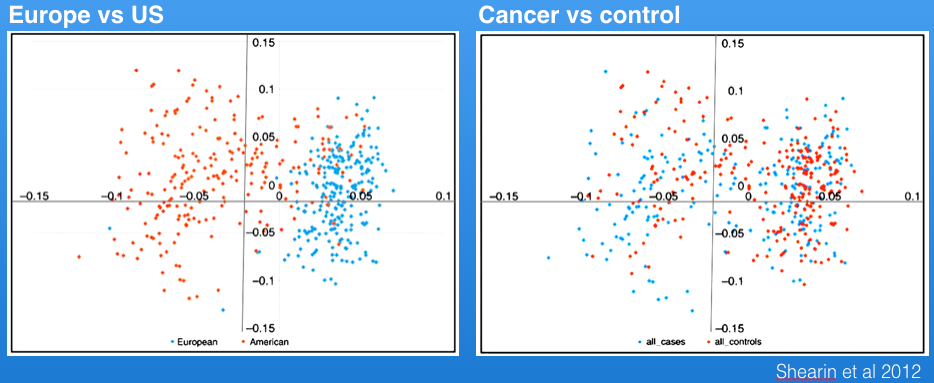
Shearin et al (2012) identified genetic structure in the Berner populations from the US vs Europe as shown in the principal components graph (left, below), but cases (affected animals) and controls were distributed across both populations.
There is a DNA histiocytic sarcoma test now being offered by Antagene. It does not identify genes that cause HS; it is an association test that identifies genetic profiles that indicate higher or lower risk of the disease. It identifies three risk classes, A, B, and C.

Optigen (US)
As you can see from the statistics below, some dogs (14%) with HS have the A profile, and some (12%) with the high risk profile, C, are healthy. This test will NOT tell you which dogs will get cancer and which won't. It tells you only that animals with profile A have a much lower risk of HS, and those with profile C are at a considerably higher risk than average.

This test is for only HS, and as indicated above HS is only one of several types of cancer in Berners. Breeders should not use this tool to remove animals from the gene pool. For example, many dogs that fall in the high risk group (C) are healthy, and there are dogs in the low risk group (A) with HS. Given the small gene pool and high level of inbreeding in Berners, it will probably be very difficult to reduce the incidence of cancer using selection.
Health Surveys
I was concerned to note this in the report for the health survey conducted in 2005 by the BMD club of America:
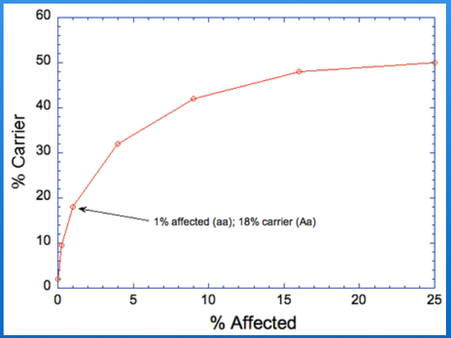
| This summary is prepared by the Health Committee of the BMDCA and represents its interpretation of the data. This summary will be organized by general information, then by organ systems for diseases with a high inidence rate, then diseases with low incidence rates and finally a summary of areas of research interests for the future. It is assumed that a disease with less than 1% incidence, 13 or fewer cases in the survey, are of low incidence and not statistically significant. |
The reason for my concern is that for a recessive mutation at an incidence of 1% affected in an "ideal" population, the expected carrier rate is not trivial - 18% (!!!). By ignoring these problems recorded at low incidence, breeders are likely to delay addressing an emerging problem until the carrier rate is 30-40%.

SUMMMARY
The Bernese Mountain Dog has significant genetic issues that need to be addressed urgently.
The average level of inbreeding of Bernese Mountain Dogs based on DNA analysis is about 35%, which is much higher than the level of inbreeding predicted from a cross of full siblings from unrelated parents, which would be 25%. On average, any two individuals in the breed are more closely related than full siblings. About 15% of the genome is homozygous and shared in every animal, which means there is no possibility of selecting for or against genes in those regions. Popular sires have played a significant role in the loss of genetic diversity in the breed.
The size of the gene pool is very small (< 20). Despite the relatively large number of animals in the breed, the fraction that are bred is small and only half as many sires are used as dams. As a result, the effective population size is low, which will result in a higher rate of increase in inbreeding than if a larger number of animals were used for breeding and the ratio of males to females was more balanced.
Genetic variation is the raw material for selection. The Berner has high inbreeding and also a large fraction of the genome is homozygous in every individual. You can't improve a trait or breed away from a problem if there is little genetic variation in the gene pool. This is a significant issue for the Berner, because breeders simply don't have the raw material - the genetic variation - that they need to improve traits and reduce disease.

The Bernese Mountain Dog faces the same problems as most other breeds. The size of the gene pool is finite and determined by the number of original founder dogs, plus any animals that were crossed in afterwards (e.g., the Newfoundland). Genes are lost from the gene pool every generation through selection and just by chance due to "genetic drift". Breeders retain the genes for type through selection, but the genes that "run the dog" - what I call the "dog genes" - can be lost, which results in health problems and lower vitality. The expression of genetic diseases increases, and while DNA tests can remove risk from known mutations, there are many mutations in the gene pool of every breed that we don't know about until they cause a problem. For this reason, DNA testing alone will not improve the overall health of dogs, but only affects a single disorder.
To improve overall health, the level of inbreeding must be reduced, and while it might be possible to make some improvements by making the best possible use of genetic variation that exists in the breed (as is evident in the graphs for genetic structure), the high rate of homozygosity and very small gene pool indicate that a "genetic rescue" program will be necessary to restore genetic diversity. This program should be based on careful evaluation of the current genetic status of the breed (using information like that presented here) and designed by population geneticists that have the tools to determine the breeding strategy that will be most effective and efficient. The Bernese Mountain Dog is in a much better position to work on improving genetic diversity than most breeds because of the excellent database of pedigree and health information maintained by Berner-Garde.
A genetic rescue program of this kind is ongoing for the Norwegian Lundehund, the Puffin dog, in which Lundehunds have been crossed with several closely related Nordic breeds. They have now produced F2 offspring, and these dogs look and act very much like the original Lundehund and show no signs of "Lundehund syndrome", a serious intestinal disorder that was often fatal. (You can read more about this exciting program here.)
REFERENCES
Abadie et al. 2009. Epidemiology, pathology, and genetics of histiocytic sarcoma in the Bernese Mountain Dog Breed. J Heredity 2009:100 (Supplement 1)
Dreger et al. 2016. Whole genome sequence, SNP chips and pedigree structure: building demographic profiles in domestic dog breeds to optimize genetic trait mapping. Disease Models & Mechanisms 9 (Dec 1). doi: 10.1242/dmm.027037
Maki 2011. Breeding for longevity in the Bernese Mountain Dog. (Seminar)
Parker et al., 20`17. Genomic analyses reveal the influence of geographic origin, migration, and hybridization on modern dog breed development. Cell Reports 19: 697-708.
Quignon et al. 2007. Canine population structure: assessment and impact of intra-breed stratification on SNP-based association studies. PLoS ONE 2(12): e1324. doi:10.1371/journal.pone.0001324.
Shannon et al. 2016. Genetic structure in village dogs reveals a Central Asian domestication origin. PNAS 112 (44): 13639-13644. doi: 10.1073/pnas.1516215112.
Shearin et al. 2012. The MTAP-CDKN2A locus confers susceptability to a naturally occurring canine cnacer. Cancer Epidemiol Biomarkers Prev. 21: 1019-1027. doi:10.1158/1055-9965.EPI-12-0190-T.
Wang et al. 2016. Merging pedigree databases to describe and compare mating practices and gene flow between pedigree dogs in France, Sweden and the UK. Anim Breed & Genetics (2016): 1-10. doi: 10.1111/jbg.12242.
Abadie et al. 2009. Epidemiology, pathology, and genetics of histiocytic sarcoma in the Bernese Mountain Dog Breed. J Heredity 2009:100 (Supplement 1)
Dreger et al. 2016. Whole genome sequence, SNP chips and pedigree structure: building demographic profiles in domestic dog breeds to optimize genetic trait mapping. Disease Models & Mechanisms 9 (Dec 1). doi: 10.1242/dmm.027037
Maki 2011. Breeding for longevity in the Bernese Mountain Dog. (Seminar)
Parker et al., 20`17. Genomic analyses reveal the influence of geographic origin, migration, and hybridization on modern dog breed development. Cell Reports 19: 697-708.
Quignon et al. 2007. Canine population structure: assessment and impact of intra-breed stratification on SNP-based association studies. PLoS ONE 2(12): e1324. doi:10.1371/journal.pone.0001324.
Shannon et al. 2016. Genetic structure in village dogs reveals a Central Asian domestication origin. PNAS 112 (44): 13639-13644. doi: 10.1073/pnas.1516215112.
Shearin et al. 2012. The MTAP-CDKN2A locus confers susceptability to a naturally occurring canine cnacer. Cancer Epidemiol Biomarkers Prev. 21: 1019-1027. doi:10.1158/1055-9965.EPI-12-0190-T.
Wang et al. 2016. Merging pedigree databases to describe and compare mating practices and gene flow between pedigree dogs in France, Sweden and the UK. Anim Breed & Genetics (2016): 1-10. doi: 10.1111/jbg.12242.
Check out
ICB's online courses
*******************
Join our Facebook Group
ICB Breeding for the Future
...the science of dog breeding
*******************
Visit our Facebook Page
ICB Institute of Canine Biology
...the latest canine news and research

